エーザイ株式会社(本社:東京都、代表執行役CEO:内藤晴夫、以下 エーザイ)とバイオジェン・インク (Nasdaq: BIIB、本社:米国マサチューセッツ州ケンブリッジ、CEO:Christopher A. Viehbacher、以下 バイオジェン)は、本日、抗アミロイドβ(Aβ)プロトフィブリル抗体レカネマブ(開発品コード:BAN2401)について、脳内アミロイド病理が確認されたアルツハイマー病(AD)による軽度認知障害(Mild Cognitive Impairment:MCI)および軽度AD(これらを総称して早期ADと定義)を対象とした大規模なグローバル臨床第Ⅲ相Clarity AD検証試験の結果を米国カリフォルニア州サンフランシスコおよびバーチャルで開催されている第15回アルツハイマー病臨床試験会議(CTAD:Clinical Trials on Alzheimer’s Disease)において発表しましたのでお知らせします。
CTADサイエンティフィック・セッションにおけるレカネマブのデータ発表の要旨
●Clarity AD試験デザイン
本試験は、北米、欧州、アジアの235施設で早期AD当事者様1,795人(レカネマブ投与群:898人、プラセボ投与群:897人)を対象とした、プラセボ対照、二重盲検、並行群間比較、無作為化グローバル臨床第Ⅲ相検証試験です。被験者は、レカネマブ投与群(10 mg/kg bi-weekly静脈投与)またはプラセボ投与群に1:1で割り付けられ、疾患ステージ(ADによるMCIまたは軽度AD)、AD症状改善薬併用の有無(例:アセチルコリンエステラーゼ阻害薬、メマンチンまたはその両方)、ApoE4ステータス、および地域によって層別割付けされました。被験者登録基準においては、多様な合併症あるいは併用治療(高血圧症、糖尿病、心疾患、肥満、腎臓病、抗凝固剤併用など)を許容しています。本試験では、民族的・人種的多様性を考慮した被験者登録を推進した結果、米国では登録被験者の4.5%が黒人、22.5%がヒスパニック系となりました。
主要評価項目は、全般臨床症状の評価指標であるCDR-SB1(Clinical Dementia Rating Sum of Boxes)の18カ月時点におけるベースラインからの変化とし、重要な副次評価項目として、アミロイドポジトロン断層法(PET)測定(センチロイド法)による脳内アミロイド蓄積、ADAS-Cog142(Alzheimer's Disease Assessment Scale-Cognitive subscale 14)、ADCOMS3(Alzheimer’s Disease Composite Score)およびADCS MCI-ADL4(Alzheimer's Disease Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment)の投与18カ月時点でのベースラインからの変化が設定されました。また、タウPETで測定する脳内タウ病理(n=257)、脳脊髄液(CSF)バイオマーカー(n=281)を評価しました。
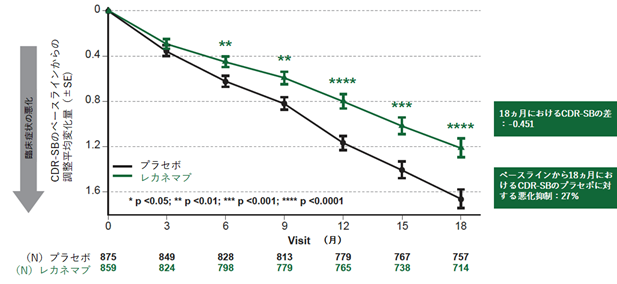
●Clarity AD試験の有効性評価結果
主要評価項目である投与18カ月時点のCDR-SBスコアのベースラインからの平均変化量について、レカネマブ投与群、プラセボ投与群はそれぞれ1.21、1.66であり、その変化量の差は-0.45([95%信頼区間(CI): -0.67, -0.23];P = 0.00005)となり統計学的に高度に有意な結果が認められ、レカネマブ投与群はプラセボ投与群と比較して27%の全般臨床症状の悪化抑制を示しました。CDR-SBスコアの平均変化量の差は、投与6カ月時点(変化量の差:-0.17 [95%CI:-0.29, -0.05];P<0.01)から3カ月毎のすべての評価時点においてプラセボ投与群と比較して統計学的に高度に有意な結果を示し、その絶対値は経時的に拡大を示しました(全評価ポイントでp<0.01)(図1)。
また、すべての重要な副次評価項目において、レカネマブ投与群はプラセボ投与群と比較して統計学的に高度に有意な結果(P<0.001)が認められました。アミロイドPET評価では、レカネマブ投与後3カ月からすべての評価時点で、統計学的に有意な脳内アミロイド蓄積の減少がみられ、投与18カ月時点のレカネマブ投与群における脳内アミロイドの平均変化量(センチロイド)は-55.5、プラセボ投与群で3.6(平均差:-59.1 [95% CI:-62.6, -55.6]; P<0.00001 )でした。投与18カ月時点でのADAS-Cog14評価では26%の認知機能低下の抑制(平均差: -1.44[95%CI:-2.27, -0.61];P=0.00065 )、ADCOMSでは24%の疾患進行の抑制(平均差:-0.050[95%CI:-0.074, -0.027];P=0.00002)、ADCS MCI-ADLでは37%の日常生活動作低下の抑制(平均差:2.016 [95%CI:1.208, 2.823];P<0.00001)を示しました。さらに、主要な層別解析では、疾患ステージ(ADによるMCIまたは軽度AD)、ApoE4ステータス(非保持者、保持者)、AD症状改善薬併用の有無、および地域(北米、アジア、欧州)のいずれのサブグループにおいても、レカネマブ投与18カ月時点のCDR-SB、ADAS-Cog14、ADCS MCI-ADLについて一貫した悪化抑制が認めらました。